

Die Verantwortliche Person MDR Artikel 15 ist dafür verantwortlich, die Konformität von Medizinprodukten mit den Anforderungen der MDR (Medical Device Regulation) sicherzustellen. Im MDR wird diese verantwortliche Person - Person Responsible for Regulatory Compliance (PRRC) - genauso gefordert wie im IVDR.
Nachfolgend erfahren Sie mehr zu den PRRC Aufgaben und Kompetenzen. Dabei können Sie sich auch weitergehend zur Konformitätsbewertung und technischen Dokumentation informieren. Zudem erläutern wir Ihnen, was in Artikel 5 des MDR steht und inwiefern sich die verantwortliche Person MDR vom Sicherheitsbeauftragten in der Medizinprodukteindustrie unterscheidet.
Welche Aufgaben hat die verantwortliche Person MDR?
Zu den Aufgaben der verantwortlichen Person MDR (PRRC Aufgaben) gemäß Artikel 15 der Medical Device Regulation gehört wie gesagt, dass die verantwortliche Person MDR sicherstellt, das Medizinprodukte den Anforderungen der MDR entsprechen. Dies sind einige ihrer wichtigsten Aufgaben:
- Konformitätsbewertungsverfahren: Zu den Aufgaben der Verantwortlichen Person MDR gehört es, Konformitätsbewertungsverfahren gemäß Anhang IX der MDR durchzuführen oder durchführen zu lassen. So gewährleistet sie, dass das Medizinprodukt den Anforderungen der MDR entspricht.
- Technische Dokumentation: Die technische Dokumentation des Medizinprodukts ist ebenfalls ein wichtiger Teil der PRRC Aufgaben. Diese Dokumentation muss alle relevanten Informationen über das Medizinprodukt enthalten. Hierzu zählen seine Eigenschaften, Konstruktions- und Herstellungsprozesse, Leistung und Sicherheit sowie die klinische Bewertung.
- Überwachung der Konformität - Vigilanz: Zu den Pflichten der Person Responsible for Regulatory Compliance gehört es auch, die Konformität des Medizinprodukts während seiner gesamten Lebensdauer zu überwachen (Vigilanz). Dies umfasst die Überwachung von Produktrückrufen, Meldungen von schwerwiegenden Vorkommnissen, der Überwachung der Sicherheit und Leistung des Medizinprodukts, sowie die Umsetzung von Maßnahmen zur Korrektur von Abweichungen. Hierzu soll der Medizinproduktehersteller ein ein Vigilanz-System implementieren.
- Zusammenarbeit mit Behörden: Eine weitere PRRC Aufgabe ist die enge Zusammenarbeit mit den zuständigen nationalen Behörden. Dies dient ebenfalls der Sicherstellung der Konformität des Medizinprodukts mit den Anforderungen der MDR. Sie muss z.B. die Behörden über schwerwiegende Vorkommnisse und andere Probleme im Zusammenhang mit dem Medizinprodukt informieren.
- Berichtspflichten gemäß den Artikeln 87 bis 91
- Benennung von Bevollmächtigten: Wenn die Verantwortliche Person nicht in der EU ansässig ist, muss sie eine bevollmächtigte Person in der EU benennen, die ihre Aufgaben im Zusammenhang mit der Konformitätsbewertung und Überwachung des Medizinprodukts erfüllt.
Insgesamt ist die Verantwortliche Person eine Schlüsselfigur bei der Umsetzung der MDR und trägt wesentlich dazu bei, die Sicherheit und Leistung von Medizinprodukten zu gewährleisten.


Weitere passende Blogbeiträge zum Thema:
Entdecken Sie auch unsere anderen Blogbeiträge zum Qualitätsmanagement für Medizinprodukte oder zum Audit und erhalten Sie Expertenwissen unter anderem zu diesen Themen:
Was ist ein Konformitätsbewertungsverfahren gemäß MDR?
Ein Konformitätsbewertungsverfahren ist ein Verfahren gemäß der Medical Device Regulation (MDR), das ein Hersteller durchführen muss, um sicherzustellen, dass sein Medizinprodukt den Anforderungen der MDR entspricht und daher auf dem europäischen Markt zugelassen werden kann.
Das Konformitätsbewertungsverfahren ist ein mehrstufiger Prozess. Dieser Prozess hängt vom Risiko des Produkts ab und kann unterschiedliche Schritte umfassen. Hierzu zählt etwa die Konformitätsbewertung durch den Hersteller selbst. Bei niedrigen Risikoklassen kann der Medizinproduktehersteller das Verfahren selbst durchführen. Dabei stellt er eine Konformitätserklärung aus, in der er bestätigt, dass das Medizinprodukt den geltenden Anforderungen entspricht. Bei höheren Risikoklassen muss eine benannte Stelle, eine unabhängige Einrichtung, die von der Europäischen Kommission benannt wurde, hinzugezogen werden. Die benannte Stelle bewertet das Medizinprodukt und überprüft, ob es den Anforderungen der MDR entspricht. Sie stellt dann ein Konformitätsbewertungszertifikat aus, das den Hersteller berechtigt, das Medizinprodukt auf dem europäischen Markt zu vertreiben.
Für einige Medizinprodukte, insbesondere solche mit höherem Risiko, erfordern eine klinische Bewertung, um die Sicherheit und Wirksamkeit zu bewerten. Eine klinische Bewertung beinhaltet die Bewertung der vorhandenen klinischen Daten sowie ggf. die Durchführung klinischer Studien. Nach dem Inverkehrbringen des Medizinprodukts muss der Hersteller zudem eine kontinuierliche Überwachung des Produkts durchführen (Vigilanz). Somit stellt er sicher, dass es weiterhin den Anforderungen der MDR entspricht.
Insgesamt ist das Konformitätsbewertungsverfahren ein wichtiger Prozess, um sicherzustellen, dass Medizinprodukte auf dem europäischen Markt sicher und wirksam sind und den Anforderungen der MDR entsprechen.
Interessante Produkte für Sie
Kursformen
Zertifikat
Informationen






Was bedeutet Technische Dokumentation gemäß MDR?
Die technische Dokumentation gemäß der Medical Device Regulation ist eine umfassende Sammlung von Unterlagen und Informationen, die der Hersteller eines Medizinprodukts bereitstellen muss, um die Konformität seines Produkts mit den Anforderungen der MDR nachzuweisen. Die technische Dokumentation umfasst dabei alle relevanten Informationen, die für die Bewertung der Sicherheit, der Leistung und der klinischen Eigenschaften des Medizinprodukts erforderlich sind. Zu den typischen Inhalten der technischen Dokumentation gehören:
- Eine Beschreibung des Medizinprodukts, einschließlich seines Zwecks, seiner Zusammensetzung, seiner Funktionen und seiner Anwendung.
- Eine Beschreibung des Herstellungsprozesses, einschließlich der Qualitätssicherungsmaßnahmen, die der Hersteller implementiert hat, um die Konformität des Produkts mit den Anforderungen der MDR sicherzustellen.
- Eine Zusammenfassung der Ergebnisse der Konformitätsbewertung, inklusive der Bewertung der Risiken, der Leistung und der klinischen Eigenschaften des Produkts.
- Eine Liste der angewandten harmonisierten Normen und ggf. der Verweise auf alternative Methoden oder Konzepte, die der Hersteller verwendet hat, um die Konformität seines Produkts mit den Anforderungen der MDR zu belegen.
- Informationen zur Verpackung, Kennzeichnung und Gebrauchsanweisung des Medizinprodukts.
- Eine Zusammenfassung der klinischen Daten, die zur Bewertung der Sicherheit und Wirksamkeit des Medizinprodukts verwendet wurden, einschließlich der Ergebnisse klinischer Studien und anderer relevanten Informationen.
Die technische Dokumentation ist ein wichtiger Bestandteil des Konformitätsbewertungsverfahrens gemäß der MDR. Zuständige Behörden überprüfen diese, um die Konformität des Medizinprodukts zu bewerten und die Sicherheit und Wirksamkeit des Produkts für die Verwendung in der Europäischen Union zu gewährleisten.
Was steht in Artikel 15 MDR?
Artikel 15 der Medical Device Regulation (MDR) legt die Anforderungen an die für die Einhaltung der Regulierungsvorschriften verantwortliche Person fest, die von Herstellern von Medizinprodukten benannt werden muss. Die Verantwortliche Person ist für die Konformitätsbewertung und die Überwachung der Konformität des Medizinprodukts mit den Anforderungen der MDR verantwortlich. Artikel 15 der MDR fordert, dass Medizinproduktehersteller die Verantwortliche Person schriftlich benennen und eine ständige Erreichbarkeit sicherstellt ist. Sie muss über die notwendigen Kompetenzen und Befugnisse verfügen, um die in Artikel 15 aufgeführten PRRC Aufgaben durchzuführen.
Zudem stellt die verantwortliche Person (Person Responsible for Regulatory Compliance) die Überwachung der Konformitätsbewertung des Medizinprodukts sicher. Damit sorgt sie dafür, dass alle notwendigen Schritte zur Einhaltung der Anforderungen der MDR eingeleitet werden. Sie stellt die regelmäßige Prüfung des Medizinprodukts sicher. Dabei arbeitet sie mit den zuständigen Behörden im Falle von Beanstandungen oder Meldungen über das Medizinprodukt zusammen. Außerdem gehört laut Artikel 15 MDR die Bereitstellung der notwendigen Informationen und Unterlagen im Zusammenhang mit dem Medizinprodukt zu den Verantwortlichkeiten. Dies schließt ebenfalls die Erstellung und Pflege des technischen Dossiers und der Durchführung der Risikobewertung mit ein. Medizinproduktehersteller müssen alle notwendigen Prozesse und Verfahren implementieren, um die Konformität des Medizinprodukts mit den Anforderungen der MDR sicherzustellen.
Insgesamt fordert Artikel 15 der MDR, dass die Verantwortliche Person über die notwendigen Kompetenzen und Befugnisse verfügt, um sicherzustellen, dass das Medizinprodukt den Anforderungen der MDR entspricht und dass alle notwendigen Schritte zur Einhaltung dieser Anforderungen unternommen werden.
Alle unsere YouTube Videos finden Sie hier auf dem YouTube Kanal der VOREST AG!
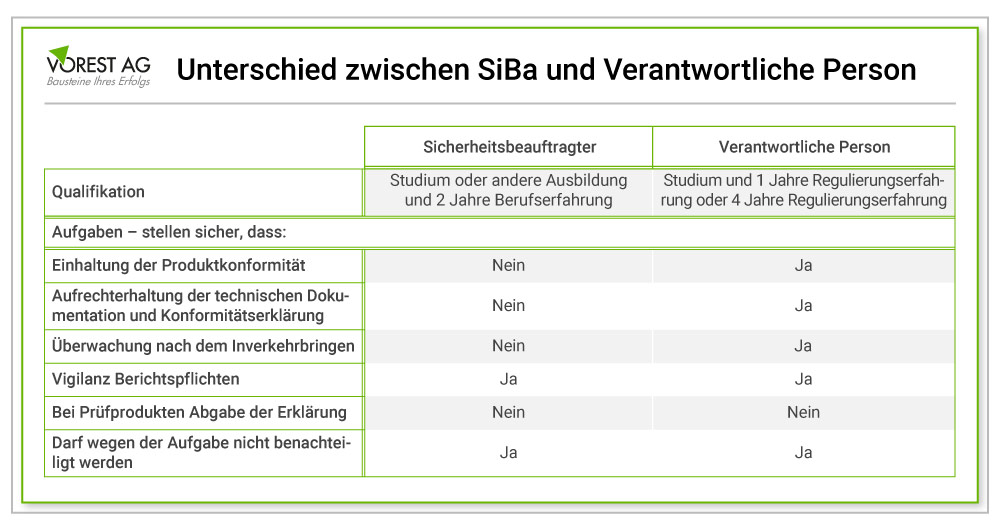
Was sind die Unterschiede zwischen Sicherheitsbeauftragter und verantwortliche Person gemäß MDR?
Gemäß der Medical Device Regulation (MDR) gibt es Unterschiede zwischen dem Sicherheitsbeauftragten und der verantwortlichen Person.
Der Sicherheitsbeauftragte (SiBa) ist eine Person, die durch die Benennung des Herstellers verantwortlich dafür ist, sicherzustellen, dass das Medizinprodukt sicher produziert, verwendet und gewartet wird. Er ist dafür verantwortlich, die Konformität des Produkts mit den einschlägigen Sicherheitsvorschriften zu gewährleisten. Zudem stellt der SiBa sicher, dass das Risikomanagement des Produkts angemessen durchgeführt wird. Der Sicherheitsbeauftragte muss über die notwendige Qualifikation und Kenntnisse verfügen, um die ihm übertragenen Aufgaben effektiv auszuführen.
Die verantwortliche Person hingegen ist eine vom Hersteller benannte Person, die die Verantwortung für die Einhaltung der MDR trägt. Sie muss sicherstellen, dass das Medizinprodukt (MP) den geltenden Bestimmungen entspricht. Das MP muss in der Lage sein, seine beabsichtigte Verwendung sicher und wirksam zu erfüllen. Die verantwortliche Person ist auch dafür verantwortlich, die technische Dokumentation des Produkts zu erstellen und aufrechtzuerhalten. Ebenfalls koordiniert sie im Falle von Produktrückrufen oder -korrekturen entsprechende Überwachungs- und Rückrufmaßnahmen.
Zusammenfassend lässt sich sagen, dass der Sicherheitsbeauftragte dafür verantwortlich ist, sicherzustellen, dass das Medizinprodukt sicher hergestellt, verwendet und gewartet wird. Die verantwortliche Person ist hingegen dafür verantwortlich, dass das Medizinprodukt den geltenden Vorschriften entspricht und seine beabsichtigte Verwendung sicher und wirksam erfüllt.
Welche Kompetenzen benötigt eine verantwortliche Person MDR?
Die Verantwortliche Person gemäß der Medical Device Regulation (MDR) muss über eine Vielzahl an Fachkenntnissen und Fähigkeiten verfügen, um den Verpflichtungen dieser Position nachzukommen. Hier sind einige der wichtigsten Kompetenzen, die eine Verantwortliche Person MDR benötigt, um die PRRC Aufgaben effektiv zu erfüllen:
- Fachwissen: Die Verantwortliche Person muss ein tiefes Verständnis für die Anforderungen der MDR und der relevanten Normen und Richtlinien haben, die für die Konformitätsbewertung und Prüfung bzw. Überwachung von Medizinprodukten gelten. Sie muss auch über ein fundiertes Fachwissen in Bezug auf die Produkte verfügen, für die sie verantwortlich ist.
- Erfahrung: Ausreichende Berufserfahrung im Bereich der Medizinprodukte ist notwendig, um die technischen und regulatorischen Aspekte ihrer Aufgaben zu verstehen. Eine mehrjährige Erfahrung in der Medizinprodukteindustrie oder in der Konformitätsbewertung von Medizinprodukten wäre ideal.
- Projektmanagement: Als PRRC müssen Sie in der Lage sein, effektiv Projekte zu planen, zu koordinieren und zu überwachen. Dies erfordert Fähigkeiten in der Budgetplanung, Ressourcenplanung, Zeitmanagement und Risikomanagement.
- Kommunikation: Sie müssen klar und präzise kommunizieren können, sowohl schriftlich als auch mündlich. Es liegt bei Ihnen, technische sowie regulatorische Informationen an ein breites Publikum zu vermitteln und entsprechende Regulierungsfragen somit zu klären. Hierzu zählen auch Kollegen, Behörden und interne sowie externe Kunden.
- Problemlösung: Die Verantwortliche Person muss in der Lage sein, komplexe technische und regulatorische Probleme zu lösen. Dazu gehört die Fähigkeit, Abweichungen zu identifizieren, Ursachen zu analysieren und wirksame Korrekturmaßnahmen zu planen und umzusetzen.
- Interpersonelle Fähigkeiten: In dieser wichtigen Position müssen Sie zur Erfüllung Ihrer Pflichten effektiv mit anderen Personen zusammenarbeiten können. Dabei gilt es auch Konflikte zu lösen, Kompromisse zu finden und Beziehungen aufzubauen.
Insgesamt erfordert die Rolle der Verantwortlichen Person MDR ein breites Spektrum an Fachkenntnissen und Fähigkeiten, um die Konformität von Medizinprodukten sicherzustellen und die Sicherheit und Leistung von Medizinprodukten zu gewährleisten.
Kostenlose Vorlage -
Erstellung einer Prozessbeschreibung

Kostenlose Vorlage -
Erstellung einer Arbeitsanweisung

Kostenloser E-Kurs -
Was ist ein Audit?

Unsere Serviceangebote im Bereich Qualitätsmanagement Medizinprodukte
- Grundlagenwissen zum Thema: Qualitätsmanagement Medizinprodukte ISO 13485 – Definition & Ziele
- Ausbildungen & Weiterbildungen: ISO 13485 Schulung - Qualitätsmanagement Medizinprodukte
- E-Learning Kurse: Medizinprodukte Online Schulungen & ISO 13485 E-Learning
- Inhouse-Training: Qualitätsmanagement Medizinprodukte Inhouse Schulungen bei Ihnen im Unternehmen
- Musterdokumente: Qualitätsmanagement Medizinprodukte Vorlagen und Checklisten
- Wissensbausteine: Expertenwissen zum Qualitätsmanagement Medizinprodukte
- Fachzeitschrift PRO SYS: Monatliche Fachinfos inklusive Musterdokumente
- Beratung: Wir unterstützen Sie beratend zum Qualitätsmanagement Medizinprodukte

Ich helfe Ihnen gerne weiter!
Kati Schäfer
Produktmanagement Training & PRO SYS
Tel.: 07231 92 23 91 - 0
E-Mail: kschaefer@vorest-ag.de